DFG founded research groups » Research Group Simon » Research
Research Group Simon
Our research interests are focused on understanding the synaptic and cellular mechanisms involved in neurodegenerative diseases, with emphasis on spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS). In particular, our group is interested in deciphering the correlation, or at times the disconnect, between neuronal death and neuronal dysfunction. We combine electrophysiology, whole tissue clearing, modern imaging, and molecular biology in mouse models of motor neuron diseases to study defects in motor circuits and their underlying mechanisms.
Mechanisms of motor unit pathology in SMA and ALS mice (DFG funded)
A motor unit consists of one motor neuron and all of the muscle fibers that it innervates. The motor unit serves as a bridge between the central and peripheral nervous system and commands movement by forming a specialized excitatory synapse between motor neuron and muscle fiber, the neuromuscular junction (NMJ). Degeneration of motor units is the hallmark of motor neuron diseases, causing muscle atrophy, paralysis and ultimately death of both SMA and ALS patients. Previously, we showed that inhibition of the p53 death pathway entirely prevents motor neuron death in SMA mice (Simon et al., 2017). However, these recued motor neurons still retract their axons from the NMJs at the muscle, implying that different mechanisms may be involved in motor neuron death and axonal pathology. Therefore, we now focus on NMJ pathology in ALS and SMA mice to understand the mechanisms of axonal retraction and its link to motor neuron dysfunction.
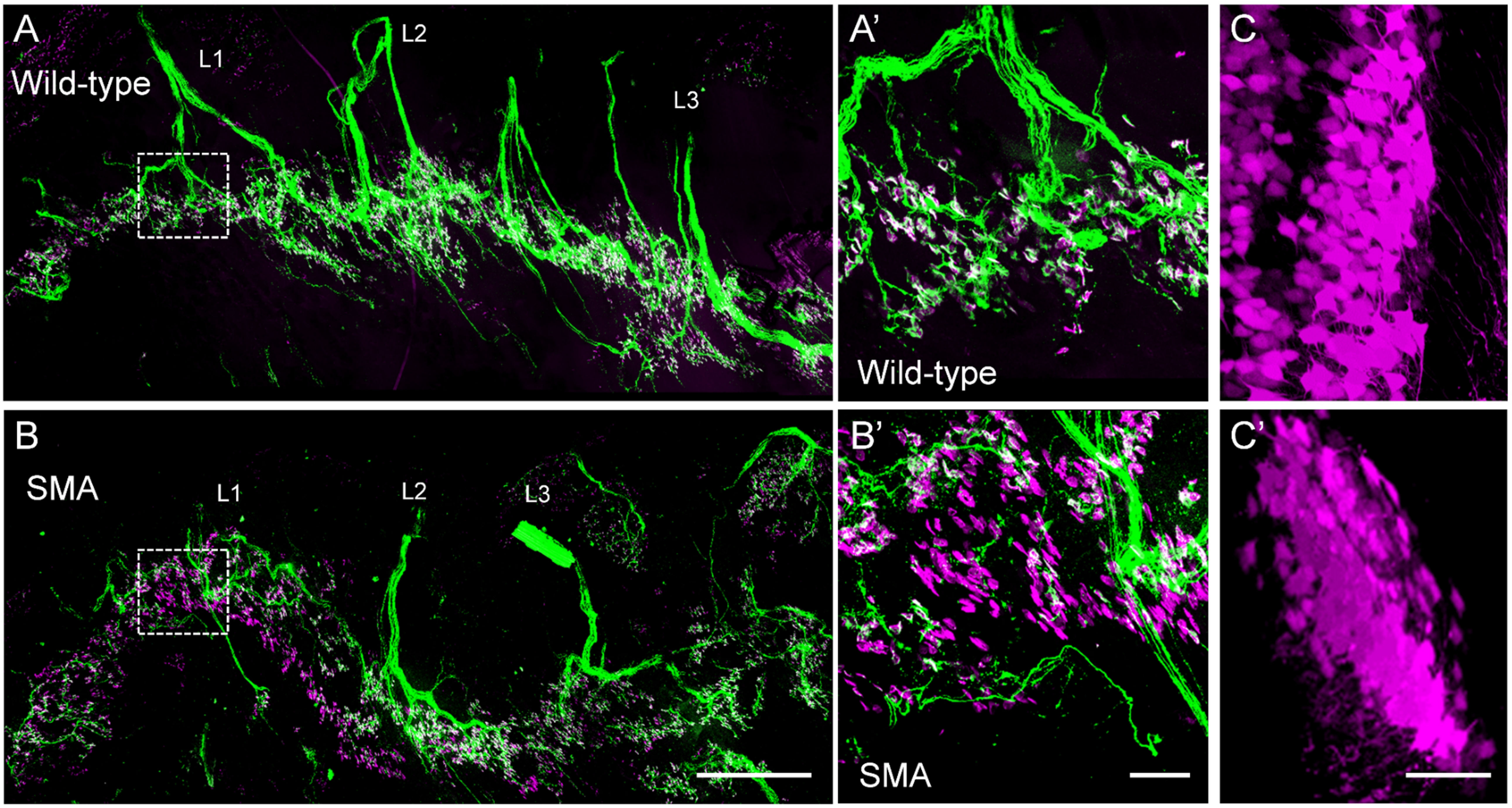
Whole tissue clearing to visualize motor units in motor neuron diseases.
Motor axons (green) innervating postsynaptic parts of the NMJ (magenta) in cleared whole mount muscle of a wild type (A) and SMA (B) animal. L1-3=lumbar ventral roots 1-3. Scale bar=300µm. NMJs innervated by L1 motor neurons are marked with dotted boxes and magnified in A’ (wild type) and B’ (SMA). Note the retraction of the motor axons from denervated NMJs in B’ (magenta, without green). Scale bar=100µm. (C) L5 motor neurons back labeled by L5 ventral root fill with a fluorescent dye, subsequent whole spinal cord clearing and (C’) 3D reconstruction. Scale bar=60µm.
Neuronal death and dysfunction in the cerebellum of ALS mice
In many neurodegenerative diseases, affected neurons become dysfunctional prior to their death. In these cases, dysfunction may be the trigger for neuronal death. To study this hypothesis, we focus on the cerebellum, in which functional and morphological changes have been detected in ALS patients. The cerebellum profoundly contributes to movement by processing sensory input to modulate spinal motor circuits. Functional impairment of the cerebellum might contribute to the impaired motor output of ALS patients. However, the mechanisms of death and dysfunction of cerebellar neurons are not well understood. To gain insight into these processes, we screen for neuronal death in cerebella of ALS mouse models in collaboration with Neil Shneider (Columbia University) using modern imaging approaches. To study functional changes in cerebellar neurons, we apply whole-cell patch-clamp recordings in a tight collaboration with Stefan Hallermann to correlate dysfunction with neuronal death in ALS in vivo. Ultimately, we plan to interfere with this disease process to test whether improving dysfunction might prevent cell death, or vice versa.
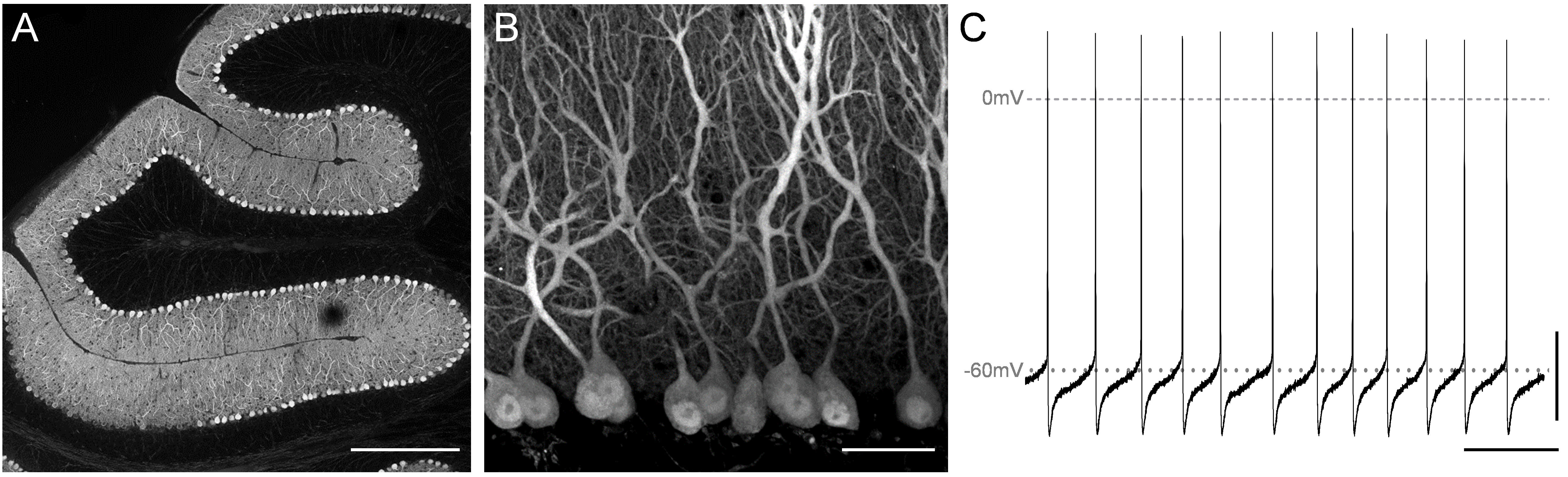 Morphological and functional characterization of cerebellar Purkinje cells.
Morphological and functional characterization of cerebellar Purkinje cells.(A) Distribution of Purkinje cells in an adult murine cerebellum. Scale bar=400µm. (B) Magnification of a small area of a cerebellar lobules reveals detailed Purkinje cells morphology. Scale bar=40 µm. (C) Spontaneous action potential firing at resting potential of a Purkinje cell using whole-cell patch clamp recording. Scale bar=20mV/50ms.